Quantumbiochemistry.org
The quantum theory applied to biology
The Hartree-Fock approximation
This method is the most basic of the methods used with electronic structure calculations. This method can be useful in qualitative and explorative research. It is also the starting point for more accurate methods which will be discussed later. Because of this it is important to understand some of the basic principles and assumptions that underlie this method.
Step 1
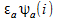
=
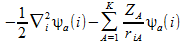 (1)
(1)
+
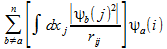 (2)
(2)
-
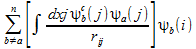 (3)
(3)
- A to K nuclei
- 1,...,i,j,...,n electrons
- a,b,...,k spin orbitals
- rij is the distance between two electrons
- riA is the distance between the ith electron and the Ath nucleus
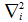 is the laplacian for the ith electron
is the laplacian for the ith electron
Step 2
We can write the electronic wave-function as a single determinant. This determinant is then called a Slater determinant.
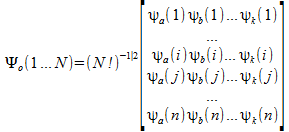
This is a Slater determinant for N electrons represented by i → k functions. ψi(i) is a one electron spin orbital. This one electron spin orbital is the product of two functions, a function that depends on the spatial (x,y,z) positions of the electron and a second function that depends on the spin of the electron. This is why we call it a spin orbital.
The spin orbitals were introduced to incorporate the Pauli exclusion principle into the wave-function. Notice that the electronic Schrödinger equation does not depend on spin, as a consequence the solution of this equation only depends on the spatial coordinates.
Step 3
The variation principle states that the energy is a functional of the ground-state wave function. We can use the slater determinant as a trail function. The slater determinant is substituted for Ψ0in equation
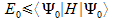
Every function ψa(i) can be represented as a linear combination of known basis functions θµ
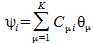
We then change the value of the expansion coefficients Cµi until we find a minimum value for E0. The resulting slater determinant is the best possible approximation to the electronic wave-function.
We impose a set of constraints on the possible solutions by using a Slater determinant as a trail function. These constraints correspond to the following assumptions:
- Every electron moves in its own orbit, ψa(i) is a one electron wave-function.
- The one electron orbitals must to be orthogonal.
- The molecular wave function vanishes when the positions (x,y,z) move to infinity.
-
The motion of the electrons with parallel spins is correlated. The use of a Slater determinant introduces terms that account for the repulsion between electrons of parallel spin. You can see this by considering the following. The probability of finding electron 1 at position 1 , electron 2 at position 2 , …, and electron n at position n can be written down like:
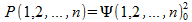 .
.
When we use the Slater determinant for wave-function
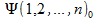
this probability is not just the product of the probabilities of finding electron 1 at position 1, electron 2 at position 2, …, electron n at position n. The expansion of the determinant will give rise to exchange terms
in the Hartree-Fock equations.
-
A Slater determinant satisfies the Pauli exclusion principle.
The Pauli exclusion Principle states that two electrons of equal spin cannot occupy the same oribtal.
When 2 electrons occupy the same spin orbital the Slater determinant vanishes.
Thus a Slater determinant is a way of imposing the Pauli exclusion principle on any possible solution of the Hartree-Fock equations.
The molecular Schrödinger equation does not include any term that refers to the Pauli exclusion principle, so the use of a Slater determinant is the only way we reckon with the Pauli exclusion principle.
-
The Hartree-Fock method assumes electrons move in the mean field of all the other electrons.
The Hartree-Fock equations are nonlinear thus we must have trial spin functions for all electrons to find the orbital energy and the spin function for a specific electron.
We start with an initial guess for the spin functions and use these guessed functions to calculate a new set of spin functions. Then we reintroduce these newly calculated spin functions into the Hartree-Fock equation to get an even better set. We then repeat this procedure until we obtain some desired convergence. This is the Self consistent field procedure (SCF procedure).
Unfortunately this assumption allows that any two electrons with opposite spin come infinitely close to one another. This is a major shortcoming of the Hartree-Fock approximation. Methods like MP2 of CI that expand on the Hartree-Fock approximation correct for this.
-
We need to make a intelligent initial guess for the one-electron orbitals.
Any function can be expanded as a linear combination of basis functions. So we expand a one-electron orbital as a linear combination of K atomic orbitals.
If the basis is complete (K → ∞) the one electron orbital will be exact.
The coefficients Cµi are numerical parameters to be found iteratively with the SCF procedure.
The functions θµ are atomic orbitals.
These functions are centered on the position of an atomic nucleus part of the molecule.
A lot of research has been done on what functions we can use as basis functions. A set of functions is called a basis set and every basis set has a code name. Many basis sets have been documented in scientific literature,these functions often are combinations of gaussion primitives.
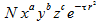
The terms a,b,c control angular momentum so that L = a+b+c with L ε {0,1,2} and ζ is a coefficient that controls the size of the atomic orbital. N is a normalization constant.
We use a computer program to solve the Hartree-Fock equations. The choice of a basis set is a trade-off between computational cost and accuracy. The more complete the basis and the larger K the higher the computational cost and the higher the accuracy. The size of the models and the accuracy of the calculations are limited by the computational speed of the hardware. Nonetheless great advances in processor speed of commercially available computers allow us to obtain a reasonable accuracy on many chemically relevant models.
-
The Rothaan-Hall and the Pople-Nesbeth equations
A set of spin orbitals can be written like:
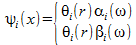 .
.
Here two electrons of opposite spin occupy the same spacial orbital. When we use electron orbitals like these we talk about the Rothaan-Hall equations. These equations can only be used to describe closed shell electron configurations because two electron are forced to reside in the same spatial orbital.
Alternatively we can write a spin orbital like:
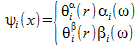 .
.
Here each electron has its own spacial orbital. When we use electron orbitals like these we talk about the Pople-Nestbeth equations. Since each electron has his own spacial orbital these equations can be used to describe open shell electron configurations. The Pople-Nesbeth equations result in one set of equations for the alpha eletrons and a second set of equations for the beta electrons. These equations can be used to represent open shell electron configurations.
Copyright© 2023 Demeester Piet